원문: 여기를 클릭하세요~
Base editors function in mouse fetuses and in the livers of adult mice to treat a genetic disorder.
The vast majority of genetic diseases are caused by single-nucleotide mutations rather than chromosomal rearrangements or small insertions or deletions (indels) and hence could potentially be therapeutically targeted by base editing. RNA-programmable deaminases, known as base editors (BEs), enable single-nucleotide cytosine-to-thymine or adenine-to-guanine conversions in a small guide RNA (sgRNA)-dependent manner in cultured cells1,2,3,4 and organisms5,6,7,8,9 and are hence well-suited to treating genetic diseases caused by single-nucleotide mutations. These BEs comprise a catalytically deficient CRISPR–Cas9 or CRISPR–CPF1 variant, derived from the two most widely used gene editing enzymes, or a Cas9 nickase variant fused to a cytosine or adenine deaminase. Unlike Cas9 or CPF1 nucleases, BEs in principle do not produce indels and do not require a donor DNA template because base editing does not rely on mutagenic nonhomologous end joining (NHEJ) or inefficient homology-directed repair (HDR) in nondividing cells, making them powerful tools for gene correction and targeted mutagenesis in vivo (Fig. 1).
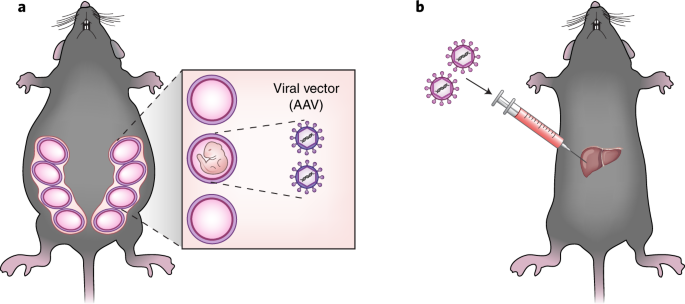
Therapeutic base editing can be achieved at various developmental stages (Fig. 1). First, BEs can be delivered into one-cell embryos or oocytes at the time of intracytoplasmic sperm injection (ICSI) to avoid mosaicism, a condition in which the organism comprises edited cells and unedited cells. Second, BEs can be delivered into fetuses before birth. Third, in vivo base editing can be achieved in newborns or adults using the appropriate vectors to deliver BEs. Recently, successful adenine or cytosine base editing has been reported in mouse embryos5,6 and in adult mice6,7. In this issue of Nature Medicine, Rossidis et al.8 and Villiger et al.9 show that cytosine BEs allow efficient base editing in mouse fetuses and in the livers of adult mice, respectively, to correct a metabolic disorder.
Base editing in early development has many advantages over that in adulthood. Genetic diseases that manifest in early developmental stages or in a whole body cannot be genetically treated in adults because treatment will be too late to have any effect or inefficient. Delivery of BEs into embryos or oocytes can achieve gene correction in every cell in newborns, avoiding mosaicism. Furthermore, embryos and fetuses are immunologically immature and therefore are unable to produce an adverse immune response against BEs or the vectors used for their expression. Fetal gene editing in utero may also lead to correction of de novo mutations, which are generated in fetuses, rather than those inherited from parents before birth; note that almost one-third of genetic diseases are caused by de novo mutations10. With the advance of next-generation sequencing technologies, de novo mutations can now be detected in the cell-free fetal DNA that circulates in the maternal blood via noninvasive prenatal diagnosis11. In utero gene editing allows for correction of de novo mutations before birth and may be ethically more acceptable than editing in the embryo, partially because edited genes in fetuses are not germline-transmissible unless germ cells are targeted.
In their study, Rossidis et al.8 first injected adenoviral vectors encoding BE3, a base editor composed of the Streptococcus pyogenes Cas9 (SpCas9) nickase, a uracil glycosylase inhibitor, and a cytosine deaminase, into embryonic day 16 mouse fetuses via the vitelline vein to disrupt the wild-type Pcsk9 or Hpd gene through creation of a premature stop codon. Pcsk9 encodes a key regulatory protein controlling cholesterol levels, which are reduced following its inactivation. Hpd encodes a metabolic enzyme in the tyrosine catabolic pathway. Hpd knockout can rescue the lethal hereditary tyrosinemia type 1 (HT1) phenotype, caused by a mutation in Fah which encodes another enzyme in the tyrosine catabolic pathway. The authors chose adenoviral vectors to target BE3 genes to the liver but recognized the limitations of adenoviral vectors for clinical applications because of their ability to trigger adverse immune responses in the host. As expected, point mutations were detected in the livers of newborns at frequencies of 10–15%, but they were not detected in other organs. Notably, edited alleles remained stable 3 months after birth. In contrast, mice treated at 5 weeks of age with the same adenovirus showed high frequencies of edited alleles at birth but low frequencies 3 months later. As expected, higher levels of antibodies against adenovirus and SpCas9 were detected in postnatal recipients than in prenatal recipients, confirming that the neonatal stage is more suited to base editing. Furthermore, in utero disruption of Pcsk9 resulted in reduction of cholesterol levels in mice 3 months after birth, whereas Hpd disruption rescued the lethal phenotype of HT1 in mice with a mutation in the Fahgene. Although Rossidis et al.8 disrupted two wild-type genes in this study, in utero base editing in principle can also achieve correction of inherited or de novo mutations in fetuses.
Villiger et al.9 used adeno-associated viral (AAV) vectors to deliver BE3 composed of S. aureus Cas9 (SaBE3)12, a smaller Cas9 ortholog, in lieu of SpCas9 into the liver of adult mice harboring a point mutation in the Pah gene encoding phenylalanine hydroxylase. These mice are a model for phenylketonuria, a human metabolic liver disease. Unlike adenoviral vectors, AAVs do not cause adverse immune responses and hence are more wildly used for gene therapy. Because a single AAV vector cannot encode the SaBE3 gene owing to its limited cargo size of ~4.7 kg bp, the authors developed a method to split the gene in a manner such that it will be fully assembled once the two parts are co-infected. Intravenous injection of the dual AAV8 vectors caused correction of the mutation in the liver, leading to restoration of physiological levels of phenylalanine in the blood. Remarkably, the Pah mRNA isolated from liver extracts at 14 weeks postinjection had alterations that result in a wild-type gene at frequencies of up to 63%, and there was no evidence of off-target base editing.
Programmable nucleases, including CRISPR–Cas9, zinc-finger nucleases, and transcription activator–like effector nucleases (TALENs), are now under clinical investigation in the United States, European Union, and China for the treatment of genetic diseases and various forms of cancer. Typically, target genes are disrupted rather than corrected by programmable nucleases in these clinical studies. In addition to disrupting target genes by incorporating a premature stop codon, BEs enable gene correction even in nondividing cells6 without triggering error-prone NHEJ, potentially expanding therapeutic gene editing to a new dimension. Although delivery of BEs into target cells and organs in vivo or in utero remains challenging and requires further development, it is encouraging that several groups have achieved efficient base editing in mice. Programmable deaminases are now poised to follow in the footsteps of programmable nucleases, making the entire human genome a potential drug target.