(원문)
In 1968, a defect in DNA repair was found to underlie a disorder that makes people extremely sensitive to sunlight. This finding continues to influence research into the origins, diagnosis and treatment of cancer.
Some people are born with exceptional sensitivity to sunlight. Fifty years ago, writing in Nature, the biologist James Cleaver1 reported a study of one such condition, and concluded that a failure of DNA repair was related to the extreme susceptibility of affected individuals to skin cancer. This was the first description of defective DNA repair in a genetically inherited disorder that makes people prone to cancer. The concepts that developed from this work now permeate research into the genetic origins of cancer and its treatment.
Starting in the 1870s, the Viennese dermatologist Moritz Kaposi performed pioneering work that defined a rare disorder characterized by high sensitivity to sunlight. Young patients were severely burned by brief exposure to the sun and acquired frequent skin lesions, and some had a high incidence of skin tumours. Kaposi dubbed the condition xeroderma pigmentosum2 (XP), using the Greek words for dry, pigmented skin — one of the symptoms of the disease. He recognized that this was a hereditary syndrome, but the underlying cause was not obvious.
Little research into XP was then done until the 1960s, when a process called nucleotide excision repair was discovered in bacteria3–5. In this process, enzymes clip out segments of DNA that have been damaged by light and replace them with fresh, undamaged DNA. Mutant bacterial strains were isolated that could be killed by low doses of ultraviolet radiation, and some of these were found to be unable to carry out excision repair4,5.
These concepts of DNA repair were then extended to human cells. By 1964, the biologists Robert Painter and Ronald Rasmussen had discovered that UV irradiation of mammalian cells led to a phenomenon that they interpreted as excision repair6. In their experiments, cultured human cells were supplied with radioactive molecules (bases) that could be incorporated into DNA. The cells were observed to incorporate new bases after UV irradiation, even when they were not duplicating their genomes, indicating that UV-damaged DNA was being replaced.
In 1967, Cleaver joined Painter’s laboratory in San Francisco as a postdoctoral fellow. Cleaver had obtained his PhD at the University of Cambridge, UK, where he had been using radioactive bases to label DNA in human cells. In April of that year, Cleaver read a newspaper article in the San Francisco Chronicle that mentioned research showing that skin cells grown from patients with XP were extraordinarily sensitive to UV radiation7. Cleaver raised with Painter the idea that XP might involve a mutation that causes DNA repair to be defective, and suggested investigating this possibility. Painter replied: “It’s a crazy idea, but at your stage what have you got to lose!”8
Cleaver acquired cultures of growing skin cells from people with XP, and applied newly developed techniques3,6 to determine whether the cells were capable of excision repair. The results clearly showed that DNA repair was defective in XP cells that had been damaged by UV irradiation (Fig. 1). Painter was a generous mentor, and encouraged his junior colleague to pursue this major discovery independently. Cleaver’s results were published in Nature on 18 May 1968.
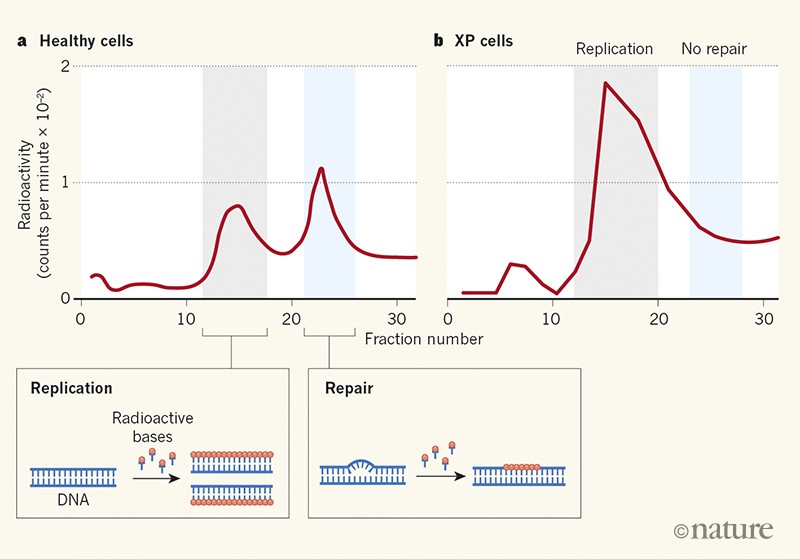
Figure 1 | Evidence of defective DNA repair in cells from people with xeroderma pigmentosum. People born with the condition known as xeroderma pigmentosum (XP) are extremely sensitive to sunlight and are prone to skin cancer. In 1968, Cleaver1 reported experiments in which cultured cells from a healthy individual and from people with XP were irradiated with ultraviolet light to cause DNA damage and then analysed to see whether the cells incorporated radioactive molecules (bases) into their DNA. a, For the healthy cells, plots of measured radioactivity for different fractions of DNA revealed distinct peaks associated with DNA replication and DNA repair. b, By contrast, the XP cells lacked the repair-associated peak. This was the first evidence that defective DNA repair underpins a genetically inherited disorder that makes people susceptible to cancer.
The paper’s conclusions were strong. Cleaver used two completely different methods to show that DNA repair in XP cells is defective, using cells from three patients clinically verified to have XP, and control cells taken from a patient with an unrelated hereditary disorder and from a healthy individual. The results suggested that XP is not a homogeneous disease, because cell lines from different individuals exhibited different levels of DNA repair. There was no indication, however, of which step was affected in the repair process, or which genes might be altered. Cleaver estimated that about 70 DNA bases were incorporated in each repair event — not far from the actual number of about 30 bases per repair event obtained later using more-precise methods9,10.
The publication generated immediate excitement. DNA repair had previously been considered a somewhat obscure topic, but Cleaver showed that it had a key role in human health. The Nobel-prizewinning molecular biologist Joshua Lederberg penned an editorial in The Washington Post highlighting this important example of fundamental research that turned out to be relevant to disease11. J. Michael Bishop, who won a Nobel prize in 1989 for his work on oncogenes, which have the potential to cause cancer, was also influenced by the finding. He wrote8: “While I was still in medical school, James Cleaver recognized xeroderma pigmentosum as a deficiency in the repair of DNA damage caused by ultraviolet light… I have been a believer in the somatic mutation hypothesis of cancer ever since”. Somatic mutations are caused by DNA damage and copying errors in the genes of tumour cells as cancer progresses. Cleaver’s paper helped to stimulate the worldwide explosion of DNA-repair research that started in the 1970s8.
Cleaver’s results were soon confirmed and extended by laboratories around the world. In 1972, it was reported that XP is a genetically complex disease12, and it is now known that alterations in eight different genes can give rise to it13,14. Seven of these genes encode components of the molecular machinery that performs excision repair; this machinery was biochemically reconstituted in vitro in the 1990s15,16. One form of XP, however, is caused by abnormal DNA synthesis after UV irradiation13, rather than by a problem in excision repair.
Specific defects in DNA repair are now known to be associated with major neurological and developmental abnormalities in other UV-sensitivity disorders, including Cockayne syndrome13,14. More broadly, it has become clear that many of the XP-associated genes have functions in addition to excision repair, and several are essential for life13,14. This means that only mild disablement of the functions of some XP genes can be tolerated.
Although XP is a rare disease (fewer than 1 person in 250,000 is affected in the United States and Western Europe)13, the consequences of mutations in XP genes are being explored widely. For example, a recent analysis found that mutations in the XPD gene (also known as ERCC2) are fairly frequent in cancer and might modulate individual responses to treatment17. There is also active research aimed at suppressing the action of XP proteins in tumour cells, to improve the effectiveness of chemotherapies that damage DNA18.
There is still no cure for XP, but intensive research into the disease means that an early diagnosis can be made. People with XP can then be protected rigorously from sunlight, allowing them a greater quality of life and longer life expectancy than was previously possible. XP societies in the United States and Europe provide support for affected children, with retreats such as Camp Sundown and Owl Patrol. Retinoid compounds can reduce the incidence of skin tumours14, and dietary interventions might improve the prospects for people with XP and related disorders19. More broadly, Cleaver’s discovery of the DNA-repair defect in XP continues to spawn vigorous research into responses to environmental DNA damage that applies not only to humans, but to every organism on the planet.
Nature 557, 648-649 (2018)