(원문)
Cycloaddition reactions are powerful tools for synthesizing three-dimensional molecules, but their scope has been limited. A creative solution to this problem opens up opportunities for drug discovery.
Reactions known as cycloadditions are unparalleled in their ability to construct ring-containing molecules in a way that precisely controls the geometric arrangement of groups attached to the carbon atoms in the molecules — that is, the reactions offer great stereoselectivity. The power of these reactions has been demonstrated in numerous syntheses of complex natural products1,2. However, the scope of cycloadditions is limited to certain combinations of starting materials, which has restricted their use for making libraries of compounds in drug-discovery programs3. Writing in Nature, Chen et al.4 report a strategy that combines cycloadditions with another type of reaction, known as carbon–carbon cross coupling, to enable the modular and programmable preparation of cycloaddition-derived molecules.
Carbon–carbon (C–C) cross-coupling reactions are often used to form bonds between carbon atoms that are already part of a carbon–carbon double bond; such carbon atoms are said to have sp2orbital hybridization. Cross couplings between sp2 carbons lend themselves to the modular synthetic routes used to make compound libraries. This has resulted in the predomination of sp2-rich structures — which tend to be two-dimensional — in compounds tested for drug discovery. But 3D molecular structures can interact with biological targets in different ways from 2D ones, so are just as important for drug discovery. Three-dimensional molecules tend to be rich in sp3 carbons, which have a different orbital hybridization from sp2 carbons, and have the capacity to form four single bonds.
A cycloaddition known as the Diels–Alder reaction provides one of the most efficient means of building sp3-rich ring systems. However, to achieve high reaction yields, stereoselectivity and regioselectivity (a preference to react at particular atoms) in Diels–Alder reactions, the electronic properties of the reactants should match. For example, in conventional Diels–Alder reactions, one of the reactants (known as a diene) should be electron-rich, whereas the other (the dienophile) should be electron-poor. This drastically reduces the number and diversity of Diels–Alder products that can be made. Chemistry students excitedly studying Diels–Alder reactions are often frustrated when they realize the restrictions involved. The constraints also prevent these reactions from being used in modular synthetic routes. Diels–Alder reactions have therefore been used much less often for drug discovery than have cross-coupling reactions3, and so their advantages for controlling the stereochemistry (the geometric arrangement of groups) of ring-containing molecules have not been fully exploited by medicinal chemists.
Workers from the same group as Chen et al. previously developed a type of cross-coupling reaction known as radical cross-coupling (RCC)5–9. This is a useful tool for converting carboxylic acids (compounds that contain CO2H groups) into products that contain alkyl, alkenyl, alkynyl or aryl groups (hydrocarbon groups that represent all the possible bonding geometries of carbon atoms). In the current paper, Chen and colleagues use carboxylic acids as a link that allows them to combine RCC and Diels–Alder reactions. The connection can be made because compounds known as anhydrides and esters serve as electronically favourable dienophiles in Diels–Alder reactions, and can then be converted into acids to take part in various RCC reactions.
On the basis of this strategy, the authors devised a simple, modular, five-step sequence to generate molecules that have 3D structural complexity (Fig. 1). In the first step, a Diels–Alder reaction involving an anhydride or ester builds a 3D molecular scaffold. This is followed by a ‘desymmetrization’ reaction10, which generates a carboxylic acid and sets the absolute stereochemistry in the resulting product. In the third step, an RCC reaction replaces the acid group with a molecular appendage, producing an intermediate that is then hydrolysed to generate a second carboxylic acid. This is used in the final step: another RCC reaction, which introduces a second appendage.
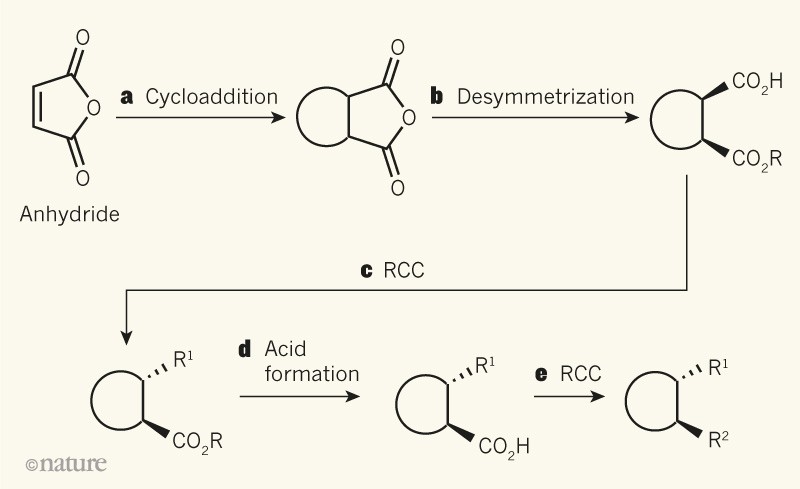
Figure 1 | A powerful strategy for making 3D molecules. Chen et al.4 report a practically simple procedure that allows access to products that cannot be made easily and directly using reactions known as cycloadditions. a, An anhydride starting material undergoes a cycloaddition to form a 3D scaffold. The ring represents several different ring structures. b, A desymmetrization reaction then forms an intermediate that contains a carboxylic acid (CO2H), in which the absolute stereochemistry (the geometric arrangement of groups) is fixed. Solid wedge bonds project above the plane of the page; R is methyl, benzyl or CH2CH2Si(CH3)3, and Si is silicon. c, The acid is replaced by a molecular appendage (R1) in a radical cross-coupling (RCC) reaction. Broken wedges project below the plane of the page. d, e, A second acid group is generated (d), and is converted to a different appendage (R2) in another RCC reaction (e).
The two RCC steps allow Diels–Alder-type products to be made that couldn’t be synthesized directly in a Diels–Alder reaction because the starting materials would have mismatched electronic properties. Another remarkable feature of this sequence is the clever mechanism of stereocontrol: the first appendage is attached to the molecular scaffold at an orientation that is governed by a nearby group produced during the desymmetrization reaction, and the orientation of the second appendage is governed by the orientation of the first. The final product therefore comprises mostly one isomer in which the two appendages are fixed in what is known as a trans orientation to each other.
Chen et al. went on to extend this chemistry from Diels–Alder reactions to three other types of cycloaddition reaction that construct rings formed of three, four or five atoms. Moreover, one of the RCC steps could be replaced with a reaction that allowed the formation of a carbon–nitrogen bond, rather than a C–C bond. The researchers suggest that bonds from carbon to other types of atom could also be made, to produce an even more structurally diverse set of final products.
The authors showcase their chemistry by using it to make natural products, pharmaceuticals and key intermediates used in the synthesis of such compounds. An example is the antipsychotic drug asenapine, which is usually manufactured as a mixture of two mirror-image isomers (enantiomers). One of these is more effective as a drug than the other, but is difficult to synthesize as a single enantiomer. Starting from a symmetrical anhydride molecule, Chen et al. show that their strategy can be used to make this enantiomer in a practically simple, short synthetic route and in good overall yield, offering several advantages over the previously reported synthesis11.
Chen and colleagues’ chemistry is easy to carry out in the laboratory. Our frustrated chemistry students can therefore immediately start to synthesize compounds that fall outside the conventional scope of Diels–Alder reactions described in their textbooks. All they have to do is to collect the anhydrides reported by the authors, the organic catalyst needed for the desymmetrization, the metal catalysts and the activating reagent used for RCC, and various other commercially available molecular building blocks (such as boronic acids). Some of these items will be used in every reaction sequence, and so the task would become even easier if a chemical-supplies company could market a kit that contained these items.
Chen and colleagues’ work will redefine how chemists think about synthesizing Diels–Alder-type products, and will find numerous applications for synthesizing molecules rich in sp3 carbons for drug discovery. The remaining challenges mainly concern the limitations of RCC. For example, the method can currently produce only a transarrangement of the two appendages introduced in the RCC steps; to make the other arrangement, the stereochemical outcome of the second RCC reaction would need to be changed. If such issues can be addressed, then Chen and colleagues’ reactions will become even more powerful tools for synthesis than they already are.