(원문)
Nature volume 560, pages E8–E9 (2018)
ARISING FROM H. Ma et al. Nature 548, 413–419 (2017)
(원문: 여기를 클릭하세요~)
In a recent publication, Ma et al.1 reported the correction of a heterozygous paternally inherited MYBPC3 mutation in human zygotes using CRISPR–Cas9 genome editing. We read their work with interest, especially the interpretation that the wild-type maternal allele was used as template to repair the DNA break at the mutation site. We think there are other explanations, specifically that repair of CRISPR–Cas9 single cleavage at the mutation site generates large deletions that prevent PCR amplification of the paternal chromosome, thereby giving the appearance of inter-homologous repair. Ma et al.1 did not perform experiments to exclude this possibility, and we therefore sought to test this idea in mouse embryos, which closely model the development of human zygotes. There is a Reply to this Comment by Ma, H. et al. Nature560, https://doi.org/10.1038/s41586-018-0381-y (2018).
Similar to Ma et al.1, we delivered the CRISPR reagents into zygotes using microinjection. We used Cas9 mRNA as opposed to CAS9 protein, as this approach has been shown to generate on-target mutations with high efficiency2. Guide RNAs (gRNAs) were identified for six autosomal loci. Three of the gRNAs targeted coding regions of genes that do not cause nullizygous lethality (Rsad2 (also known as viperin), Pik3r6 and Hmgcs2) and three targeted intronic or flanking regions (Neurog3 (also known as Ngn3), Foxp4 and Fzd3). Altogether, 127 founder embryos/mice were generated from zygotes microinjected with each gRNA. To assess the mutation efficiency at each locus, we used the polyacrylamide gel heteroduplex mobility assay (HMA)3 to screen 300–600 base-pair (bp) PCR products that span the gRNA target sites. The HMA detected mutations in 76 out of 127 (60%) of samples (Supplementary Information tab 1). These included 13 samples that generated considerably smaller PCR products than expected, indicating that they contain large deletions in the order of 100–300 bp (Supplementary Information tab 1). Notably, four samples failed to amplify, suggesting bialleic deletion of PCR primer-binding sites (Supplementary Information tab 1). To confirm the results of the HMA, we performed Sanger sequencing of the PCR products. As expected, mutations were detected in all HMA-positive samples (Supplementary Information tab 2). Notably, 93% of the HMA-negative samples that we sequenced contained a small indel compared with the wild-type sequence (Supplementary Information tab 3). Only one type of mutation was observed in most of these samples, which explained why they were not detected by HMA (false negatives). Taken together, these data show that mutations were generated in 98% of CRISPR–Cas9-microinjected zygotes.
Next, we screened specifically for large deletions using an approximately 1.6-kb PCR with primers that were equidistant from gRNA PAM sequences. Notably, we generated amplicons that were considerably less than 1.6 kb (indicating large deletions) in 35 samples. These low molecular mass products were not generated in the initial 300–600-bp PCR, indicating that they corresponded to deletions that encompass at least one of the (initial) PCR primer-binding sequences. Large deletion products were also identified in 9 additional samples when we performed an approximately 3.2-kb PCR on the Neurog3 and Foxp4 founders (Supplementary Information tab 4). These larger PCRs also generated amplicons from samples that failed to amplify in the initial 300–600-bp PCR, confirming that these contained large deletion alleles. Altogether, the number of samples containing detectable large deletions (more than 100 bp) was 57 out of 127 (45%; Fig. 1a, Supplementary Information tab 4), noting that some founders were mosaic and some contained more than one large deletion event. Large deletions were detected in 57% of the HMA-negative samples, indicating that these large deletions contributed to the amplification failure in the HMA false-negative samples. The remaining mutant HMA-negative samples may contain larger deletions that require further separation of PCR primers for detection, or may be homozygous for the indel mutation. Deleted sequences were confirmed by direct sequencing of gel-purified PCR products (Supplementary Information tab 5). Each gRNA generated a range of unique deletions, indicating that the process that underpins the generation of large deletions is stochastic (Fig. 1b, Supplementary Information tab 5). Notably, we observed that the orientation of the large deletions was asymmetric or unidirectional with respect to the cutting site (Fig. 1b, Supplementary Information tab 5). The size of the large deletions (after 1.6-kb PCR) ranged from 100 to 800 bp (Fig. 1b), with the largest deletion of 2.3 kb detected in a Foxp4 sample after 3.2-kb PCR (Supplementary Information tabs 4 and 5, sample Foxp4 #19).
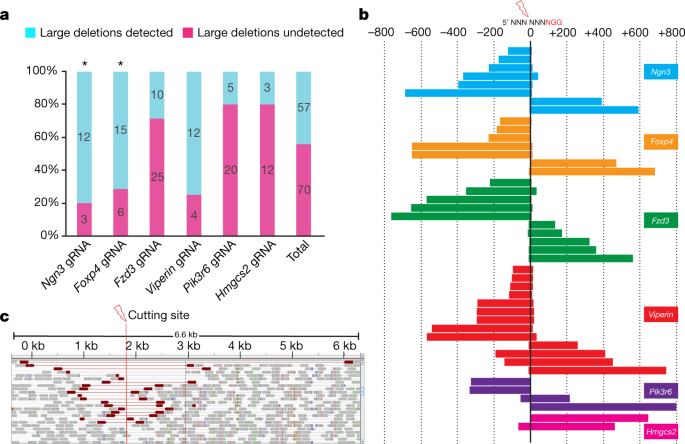
a, The number of founders that contain large deletions detected by large PCR (~1.6 kb). Asterisk symbol indicates ~3.2-kb PCR was also performed. b, Sequencing of large deletion bands after ~1.6-kb PCR from mouse zygote injection samples. ‘0’ represents the cutting site of Cas9. Each bar represents the deletion position relative to the corresponding NGG PAM sequence. c, Whole-genome sequencing analysis of Rsad2-gRNA-treated mouse ES cell pools. Integrative genome viewer snapshot shows reads paired in sequencing and sorted by insert size. Read pairs in red indicate discordantly mapping pairs indicative of large deletions.
Because the detection of large deletions using PCR is prone to amplification bias and is confounded by the deletion of primer sequences, we next sought to use an unbiased approach using PCR-free paired-end whole-genome sequencing (WGS) to determine the frequency and extent of large deletions in CRISPR–Cas9-treated mouse embryonic stem (ES) cells. We transfected ES cells with plasmid PX459.V2.04 that expresses Cas9 and Rsad2 gRNA, and puromycin-resistant transfectants were obtained for genomic DNA (gDNA) extraction and WGS analysis. Of 88 sequence reads that span the Rsad2single gRNA cleavage site, only two (2.3%) corresponded to wild-type alleles. Small indels and substitutions were found in 33 reads (37.5%), and large deletions inferred from discordant mapping of paired-end reads and split read mapping over the breakpoint were detected in 33 reads (37.5%) (Fig. 1c). The remaining 20 reads (23%) unexpectedly contained insertions of the PX459.V2.0 expression plasmid. Together, these data confirm that large deletions are frequently generated after CRISPR–Cas9-mediated DNA cleavage in mouse ES cells and zygotes.
In summary, our data demonstrate that large deletions are frequently generated in mouse zygotes after CRISPR–Cas9 single cleavage, as has recently been noted by others5,6,7. Although species differences may affect DNA repair products, Ma et al.1 cannot conclude with certainty that the purported homology-directed repair gene correction event has generated homozygous wild-type embryos until the existence of large deletions is excluded. This could be investigated by generating larger PCR products as described above. Quantitative PCR analysis to confirm the presence of both wild-type alleles would provide definitive evidence. The importance of accurate genotyping in the context of human germ-line modification cannot be overstated. Failure to detect large deletions could lead to disastrous outcomes in potential clinical applications.
Methods
gRNAs were designed using the online CRISPR tool developed by the Zhang laboratory at MIT (http://crispr.mit.edu)8. Cas9 mRNA (100 ng µl−1) and gRNAs (50 ng µl−1 each) were injected into the cytoplasm of C57BL/6N zygotes using a FemtoJet microinjector, transferred to pseudo-pregnant recipients, and allowed to develop to term or collected as embryos. gRNA sequences were as follows: Neurog3 5′-GCACAGCTGGATTCCGGACAAA-3′; Foxp4 5′-CCAGCGTTCCCATTGTCCTT-3′; Fzd3 5′-CTTAGCAAGG GTGTGAAAAG-3′; Rsad2 5′-GGGTGGCTAGATCCCGGGA-3′; Pik3r6 5′-CTTACCCTGATTGCTCTGGA-3′; and Hmgcs2 5′-TACAATCCCTCCTG CTCCCC-3′. All animal work was conducted following approval by The University of Adelaide Animal Ethics Committee in accordance with the Australian code for the care and use of animals for scientific purposes.
Data availability
All data and reagents are available from the corresponding author upon request.