(원문)
Nature Medicine volume 24, pages 899–900 (2018)
Signaling by the tumor-suppressor protein p53 antagonizes CRISPR–Cas9 gene editing of human pluripotent stem cells and immortalized human retinal pigment epithelial cells.
Now in its third decade of use1, genome editing relies on an engineered nuclease to induce a double-strand break (DSB) in a locus of interest to allow for investigator-specified genetic changes. The RNA-guided enzyme Cas9, discovered in CRISPR-type adaptive immune bacterial circuits2, is now the nuclease most widely used for this purpose. Human pluripotent stem cells (hPSCs), such as embryonic and induced pluripotent stem cells (iPSCs), were first genome-edited in the late 2000s using zinc finger nucleases (ZFNs); the literature by now has a wealth of examples of panels of edited hPSCs3. However, the efficiency of editing in this type of cell is lower than in other cell types. In this issue, Kaykas and colleagues4 and Taipale and colleagues5 discover a key role for the tumor-suppressor protein p53 in antagonizing efficient genome editing using Cas9 in hPSCs and immortalized human retinal pigment epithelial (hRPE) cells. This has important implications both for basic research and for the development of genome-edited hPSC progeny for clinical applications.
Kaykas and colleagues4 first attempted to use Cas9 to knockout 16 genes in hPSCs and found that edited cells were dying at a higher rate than their wild-type siblings; editing a gene that is silent in hPSCs produced the same result as editing one that is active. These and other data provide clear evidence that even a single DSB induced in the genome of hPSCs by Cas9 reduces their survival. Taipale and colleagues5 used immortalized human retinal pigment epithelial cells to show that in important applications of Cas9, such as screening for the effects of editing many genes in parallel, the results are confounded by a broad, gene-nonspecific effect of the DSB itself. This is conceptually similar to such an effect found in Cas9 screens in transformed cells, in which the copy number of the gene target, rather than the nature of individual genes, drives the phenotype of edited cells6. For potential clinical applications of edited hPSC progeny , this makes the path to the desired cell more difficult and potentially fraught with undesired side effects.
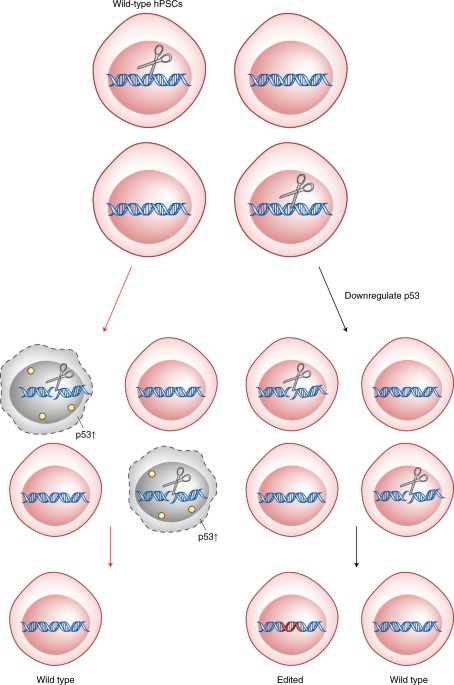
Kaykas and colleagues4 studied the transcriptional response of hPSCs to editing with Cas9 and found that the induction of a DSB by Cas9 increases expression of genes involved in programmed cell death. Furthermore, the gene whose expression was most upregulated was that for the cyclin-dependent kinase inhibitor p21; DNA damage activates this gene by signaling via p53. To provide clear evidence for the resulting hypothesis that Cas9-induced toxicity is mediated by p53, Kaykas and colleagues4 reduced the levels, or function, of p53 in the cells and found that this dramatically reduced Cas9-induced toxicity and—more importantly—increased the number of hPSCs with an edited genome that survived (Fig. 1). Taipale and colleagues5 found that their ability to place precise edits in the genome of hPSCs is antagonized by p53, and they also observed an increased efficiency of editing in hRPE cells in which p53 function was reduced.
Marina Spence/Springer Nature
Kaykas and colleagues4 show that inducing even a single DSB in the chromosomes of a pool of wild-type hPSCs causes activation of the p53 pathway and the preferential death of those cells (left). Transient suppression of p53 signaling (right) allows the edited cells to survive.
The work by these authors is of immediate use to the many practitioners of genome editing; transient suppression of p53 is well-tolerated by hPSCs, and, on the basis of the provided data, it clearly could be an option for experiments in which efficient DSB-based engineering of their genome is required. What are the clinically relevant implications of this work? Genome editing, first with ZFNs and then with TAL effector nucleases, has been used in the clinic for nearly a decade, and the safety record to date is good7,8. Two clinical trials are underway with ZFN-edited human hematopoietic stem and progenitor cells (NCT02500849 and NCT03432364), several more are imminent using Cas9, and the literature teems with examples of efficient, time-stable editing in these cells9. In contrast, no clinical trials are in progress in the United States or Europe with progeny of genome-edited hPSCs, and as of the present date, none of the five vertically integrated biotechnology companies that rely on genome editing have announced plans for such trials. That said, transplantation of differentiated progeny of human embryonic stem cells (hESCs) has entered the clinic, with promising early data for indications in the visual and cardiovascular systems10,11, and more such trials are on their way. In broad strokes, it is important to chart a path for the use of genome editing specifically in this clinical modality as well.
Kaykas and colleagues4 and Taipale and colleagues5 correctly point out that ex vivo–edited, clinic-bound hPSCs could end up experiencing selection for a mutated or downregulated p53 tumor-suppressor pathway, and this will be a first hit on the path to tumorigenesis. The application of deep-sequencing technologies has set a new level of expectation for how ‘pristine’ the genome of a transplanted cell should be, and at least one clinical trial in Japan was put on hold when a mutation was discovered that may or may not have been genotoxic. That said, it is essential to understand that subjects on clinical trials are not dosed with genomes bearing hypothetically dangerous mutations, but rather with cells. It is certain that a regulatory framework can be put in place that derisks those cells, even in settings where there is potential that a mutation may be selected for in a tumor-suppressor pathway, as has been suggested for Cas9-edited iPSCs. In this regard, an important and actionable question posed by Kaykas and colleagues4and Taipale and colleagues5 is whether transient suppression of p53 can be used to produce a genome-edited iPSC that will pass the relevant tests of preclinical safety. More generally, the findings of the authors should inspire experiments to explicitly measure and address, in the context of properly structured preclinical efforts, the associated risk, if any, of the phenomenon described. Importantly, the field of cell and gene therapy has a strong track record of addressing concerns of this type in a way that ensures continued steady progress12.